Un análisis integrado del metiloma y transcriptoma revela una fuerte desregulación del componente mieloide en el perfil epigenético de la Esclerosis Sistémica.
J. Martínez-López; L. Ortiz-Fernández; E. Estupiñán-Moreno; M. Kerick; E. Andrés-León; LC. Terrón-Camero; E. Carnero-Montoro; G. Barturen; L. Beretta; I. Almeida; PRECISESADS Clinical Consortium, ME. Alarcón-Riquelme; E. Ballestar; M. Acosta-Herrera; J. Martín.
Relevancia: Este trabajo de investigación fue publicado en la revista Arthritis & Rheumatology, con un índice de impacto de 11,4 y ubicada en el primer decil de la categoría de reumatología.
RESUMEN
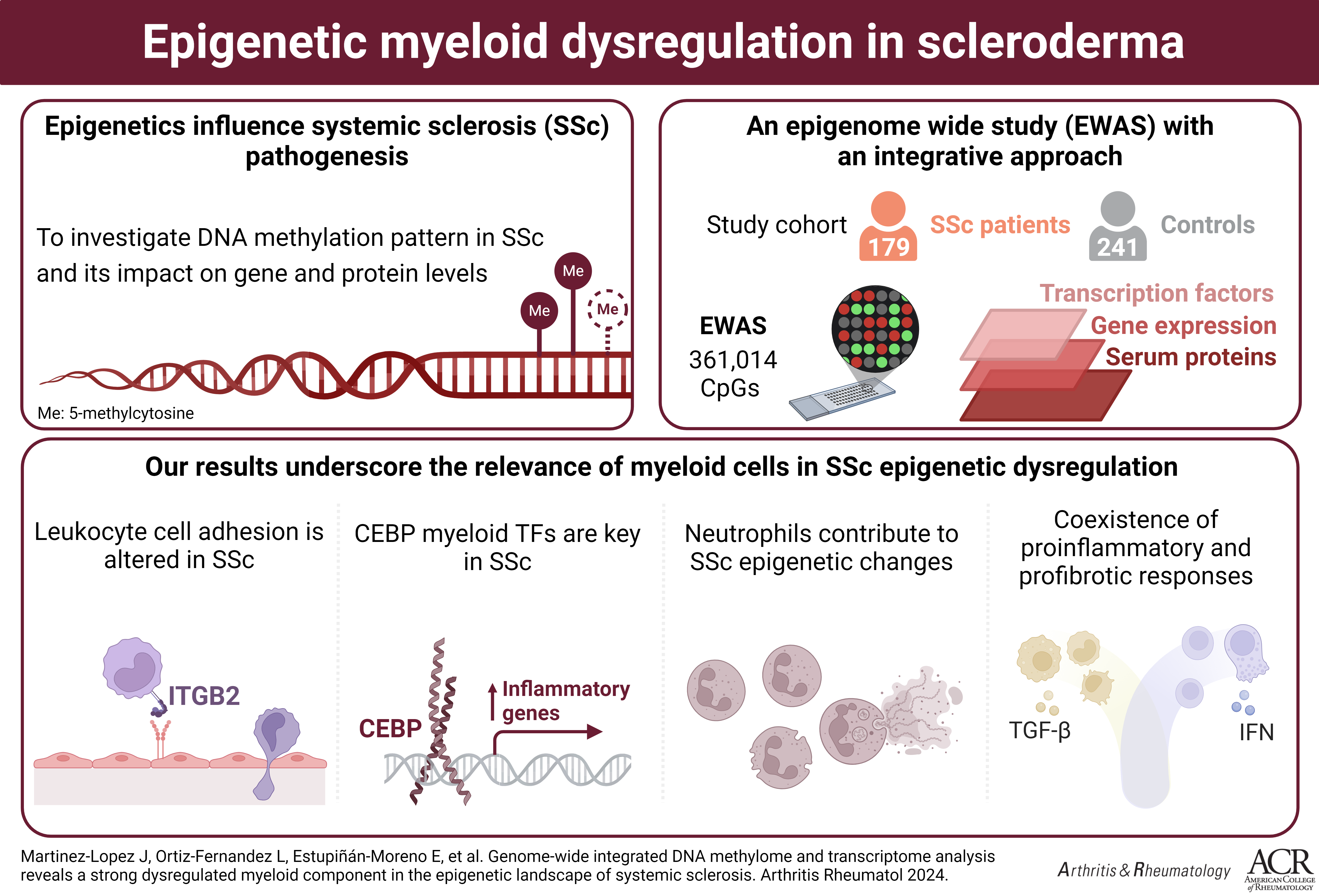
Antecedentes: El desarrollo de la esclerosis sistémica (ES), una enfermedad autoinmune sistémica, está condicionado por factores genéticos y ambientales, resaltando la importancia de la regulación epigenética en la enfermedad. Por tanto, el objetivo de este estudio es hallar cambios en la metilación del ADN asociados a la ES mediante un estudio de asociación de epigenoma completo (EWAS). Métodos: Se analizaron datos de metilación del ADN de muestras de sangre total de 179 pacientes de ES y 241 controles para identificar posiciones diferencialmente metiladas (DMPs), con una tasa de falso descubrimiento (FDR)<0.05. Estos resultados se integraron con datos de RNA-seq de los mismos pacientes para identificar el impacto funcional de esta regulación. Además, se analizó la influencia de los cambios de metilación y expresión sobre factores de transcripción y la relación entre metiloma, transcriptoma y niveles de proteínas en suero.
Resultados: Se identificaron 525 DMPs, que estaban enriquecidas en rutas relacionadas con el sistema inmunológico, siendo la adhesión leucocitaria la más significativa (FDR=4.91x10-9). Este resultado, reforzado tras el análisis de integración posterior, destaca el papel de las integrinas en la patología. Los resultados del análisis integrado de metilación y expresión muestran un enriquecimiento en rutas relacionadas con los neutrófilos, resaltando la relevancia de este tipo de célula mieloide en la patogénesis de la ES. Por último, este análisis integrado revela un incremento de la actividad de los factores de transcripción de la familia CCAAT/enhancer-binding protein (CEBP) en los pacientes de ES, moléculas esenciales del desarrollo de las células del linaje mieloide.
Conclusiones: Estos hallazgos ponen de manifiesto la desregulación epigenética presente en la patología, así como el impacto que tiene en la expresión génica, revelando nuevas moléculas con potencial aplicación clínica y avanzando en la comprensión de la patogénesis de la ES.
https://acrjournals.onlinelibrary.wiley.com/doi/10.1002/art.43044
DOI: 10.1002/art.43044